Choroby psychiczne wciąż budzą lęk i są owiane wieloma mitami. W społeczeństwie panuje przekonanie, że osoby zmagające się z depresją są „inne” lub nieprzewidywalne. Tymczasem prawda jest zupełnie inna – z chorobami psychicznymi można normalnie żyć, pracować, zakładać rodziny i spełniać marzenia. Nie są one zaraźliwe, nie świadczą o słabości, a odpowiednia terapia i leczenie farmakologiczne pomagają odzyskać równowagę.

Leczenie nie jest powodem do obaw – to szansa na poprawę jakości życia i odzyskanie kontroli nad własnym losem. Największym problemem nie jest sama choroba, ale brak wiedzy i strach przed nią. W tym artykule przybliżę temat depresji, pokazując, że jest to choroba o podłożu fizjologicznym jak inne powszechne choroby. Osoby z nią żyjące potrzebują przede wszystkim zrozumienia, a nie odrzucenia.
Depresja to nie tylko smutek czy przygnębienie. To choroba podstępna i wyniszczająca. Według Światowej Organizacji Zdrowia (WHO) depresja jest czwartą najpoważniejszą chorobą na świecie i jedną z głównych przyczyn samobójstw. Eksperci przewidują, że do 2030 roku stanie się także pierwszą najczęściej diagnozowaną jednostką chorobową na świecie. Na depresję na całym świecie choruje 350 mln ludzi, w Polsce 4 miliony.
WHO wskazuje trzy podstawowe objawy depresji:
● obniżony nastrój,
● brak radości (anhedonia),
● brak energii (anergia) oraz siedem dodatkowych:
Depresja nie jest jedynie chorobą dorosłych i zapracowanych osób – coraz częściej dotyka także dzieci i młodzież. Zgodnie z danymi Narodowego Funduszu Zdrowia, w 2021 roku około 630 tysięcy młodych ludzi wymagało specjalistycznej pomocy psychiatrycznej i psychologicznej. Warto jednak pamiętać, że w Polsce opieka psychologiczna oraz psychiatryczna finansowana przez NFZ wciąż pozostaje niewystarczająca, co sprawia, że część młodych pacjentów korzysta z prywatnej pomocy, która nie jest uwzględniana w oficjalnych statystykach. Depresja u młodych osób może się objawiać w sposób niejasny. Często zamiast smutku czy apatii występuje rozdrażnienie, pobudzenie czy agresja.
Przykładowe leki stosowane w leczeniu depresji:
1. SSRI - selektywne inhibitory wychwytu zwrotnego serotoniny
FLUOKSETYNA, CITALOPRAM, ESCITALOPRAM, PAROKSETYNA, SERTRALINA
Selektywne inhibitory wychwytu zwrotnego serotoniny (SSRI) to leki pierwszego wyboru w leczeniu depresji o nasileniu łagodnym i umiarkowanym. Stosuje się je także w terapii zaburzeń lękowych, fobii społecznej, bulimii oraz innych schorzeń związanych z dysfunkcją układu serotoninergicznego.
SSRI działają poprzez zwiększenie stężenia serotoniny w szczelinach synaptycznych, co prowadzi do odblokowania i regulacji przewodnictwa serotoninergicznego w mózgu. Są to leki o wysokim profilu bezpieczeństwa, dobrze tolerowane przez większość pacjentów.
Najczęściej występujące działania niepożądane obejmują:
● zaburzenia ze strony przewodu pokarmowego (nudności, biegunki),
● zmiany apetytu (jadłowstręt lub zwiększony apetyt),
● wpływ na funkcje seksualne, takie jak spadek libido czy zaburzenia erekcji.
SSRI nie powodują sedacji, co czyni je korzystnym wyborem dla pacjentów aktywnych zawodowo. Dodatkowo ich stosowanie może ułatwiać rozpoczęcie i prowadzenie psychoterapii, zmniejszając nasilenie objawów depresyjnych i lękowych, co sprzyja większej skuteczności terapii psychologicznej.
Ostrożność należy zachować przy jednoczesnym stosowaniu SSRI i inhibitorów MAO, ponieważ może to prowadzić do zespołu serotoninowego – potencjalnie groźnego dla życia stanu wynikającego z nadmiernego pobudzenia układu serotoninergicznego.
Zespół serotoninowy to nagły i niebezpieczny wzrost poziomu serotoniny w ośrodkowym układzie nerwowym, najczęściej spowodowany jednoczesnym przyjmowaniem leków zwiększających jej stężenie.
● Objawy neurologiczne: drżenie mięśni, pobudzenie, niepokój, splątanie, halucynacje.
● Objawy wegetatywne: wzrost ciśnienia krwi, przyspieszona akcja serca, pocenie się, gorączka.
● Objawy mięśniowe: wzmożone odruchy, sztywność mięśni, drgawki.
2. Inhibitory monoaminooksydazy (MAO)
Selektywne inhibitory MAO-A: MOKLOBEMID - odwracalnie hamuje rozpad serotoniny i dopaminy w mózgu, przez co zwiększa ich stężenie w szczelinie synaptycznej. Moklobemid jest skuteczny nawet w depresjach ciężkich, jest zasadniczo pozbawiony interakcji z lekami.
Selektywne MAO-B: SELEGILINA, RASAGILINA - stosowane w chorobie Parkinsona
Działania niepożądane: bezsenność, nudności, zaburzenia ze strony układu krążenia.
Cheese effect (efekt serowy) to potencjalnie groźna reakcja organizmu, która może wystąpić u osób przyjmujących inhibitory monoaminooksydazy (MAO) po spożyciu pokarmów bogatych w tyraminę.
Tyramina to substancja występująca w fermentowanych, dojrzewających produktach spożywczych, takich jak:
● Monoaminooksydaza (MAO) to enzym rozkładający tyraminę w organizmie.
● Inhibitory MAO blokują ten enzym, co powoduje nagromadzenie tyraminy w organizmie.
● Nadmiar tyraminy powoduje gwałtowny wzrost ciśnienia krwi, co może prowadzić do przełomu nadciśnieniowego – stanu zagrażającego życiu.
● Silny ból głowy
● Nagły wzrost ciśnienia krwi
● Pocenie się, kołatanie serca
● Nudności, wymioty
● W skrajnych przypadkach – udar mózgu lub zawał serca
1. Nieselektywne inhibitory wychwytu zwrotnego noradrenaliny i serotoniny tzw. TLPD - Trójpierścieniowe leki przeciwdepresyjne
KLOMIPRAMINA, AMITRYPTYLINA, DOKSEPINA, OPIPRAMOL
Leki te charakteryzują się wąskim indeksem terapeutycznym, co oznacza, że różnica między dawką leczniczą a toksyczną jest niewielka, zwiększając ryzyko przedawkowania. Z tego powodu ich stosowanie wymaga ostrożności i ścisłego monitorowania.
Są to leki pierwszego rzutu w depresji endogennej, a także mogą być stosowane pomocniczo w leczeniu uzależnień.
Ustępujące wraz z rozwojem tolerancji:
● Silna sedacja,
● Zaparcia, trudności w oddawaniu moczu,
● Suchość błon śluzowych, niewyraźne widzenie,
● Niepokój, majaczenie, senność,
● Zaburzenia koncentracji, upośledzenie pamięci.
Nieustępujące pomimo dłuższego stosowania:
● Przyrost masy ciała,
● Zaburzenia funkcji seksualnych,
● Ryzyko kardiotoksyczności – mogą wydłużać odstęp QT i zwiększać ryzyko arytmii.
Ze względu na powyższe działania niepożądane leki te wymagają indywidualnego dostosowania dawki oraz regularnej kontroli parametrów sercowo-naczyniowych.
2. Nieselektywne inhibitory wychwytu zwrotnego noradrenaliny i serotoniny nie zaliczane do TLPD.
WENLAFAKSYNA, DULOKSETYNA
Efekt terapeutyczny tych leków pojawia się szybciej niż w przypadku trójpierścieniowych leków przeciwdepresyjnych (TLPD). Ponadto ich profil hamowania receptorowego jest łagodniejszy, co przekłada się na lepszą tolerancję i większe bezpieczeństwo stosowania.
Wśród działań niepożądanych najczęściej występują:
● zaburzenia ze strony układu pokarmowego (nudności, biegunki, zaparcia),
● zaburzenia funkcji seksualnych, takie jak obniżone libido, opóźniony orgazm czy zaburzenia erekcji.
Leki te są stosowane głównie w depresjach o łagodnym i umiarkowanym nasileniu, a także jako środki przeciwlękowe.
Duloksetyna wyróżnia się dodatkowym wpływem na układ dopaminergiczny, co przyczynia się do poprawy napędu psychoruchowego i motywacji, co może być szczególnie korzystne u pacjentów z depresją z anhedonią i spowolnieniem.
3. Antagoniści receptorów dla monoamin
MIRTAZEPINA, MIANSERYNA, TRAZODON
Mirtazapina i mianseryna są skuteczne w depresji z bezsennością i utratą apetytu, ale mogą powodować przyrost masy ciała. Trazodon jest dobrym wyborem w depresji z lękiem i zaburzeniami snu, ale wiąże się z ryzykiem priapizmu (bolesne, długotrwałe wzwody wymagające interwencji medycznej) i hipotensji. Wszystkie trzy leki mają silne działanie nasenne, co jest korzystne w przypadku depresji z zaburzeniami snu.
4. Leki atypowe
TIANEPTYNA - blokuje receptory opioidowe, powoduje bardzo słaby wychwyt serotoniny, mimo to jest bardzo skutecznym lekiem, zwiększa syntezę BDNF, może odwracać upośledzenie poznawcze szczególnie u osób starszych. Ma dobrą tolerancję.
AGOMELATYNA - agonista receptorów dla melatoniny, zwiększa uwalnianie noradrenaliny i dopaminy, przywraca rytm dobowy.
WORTIOKSETYNA - jest skutecznym lekiem w leczeniu depresji i zaburzeń lękowych. Dzięki zwiększeniu poziomu serotoniny w mózgu, pomaga poprawić nastrój pacjentów i zmniejsza objawy związane z tymi zaburzeniami.
BUPROPION - skuteczny szczególnie w depresji sezonowej (depresja występująca podczas jesieni i zimy) oraz depresji z obniżeniem napędu. Może służyć jako augmentacja terapii SSRI lub lek stosowany w monoterapii. Działa poprzez zwiększenie stężenia dopaminy w szczelinach synaptycznych układu dopaminergicznego oraz noradrenergicznego. Może podwyższać ciśnienie oraz powodować mdłości - nasilone głównie zaraz po rozpoczęciu terapii lekowej. Lek nie powoduje przybierania na masie, nie powoduje również dysfunkcji seksualnych. Może powodować spadek masy u pacjentów, dlatego nie jest lekiem z wyboru u osób zmagających się z zaburzeniami odżywiania.
Dlaczego ludzie chorują na depresję?
W tej części artykułu omówię hipotetyczne przyczyny depresji, wyjaśniając, czym jest ta choroba oraz dlaczego ma ona podłoże fizjologiczne, co sprawia, że wymaga leczenia, tak jak każda inna choroba.
1. HIPOTEZA MONOAMINERGICZNA
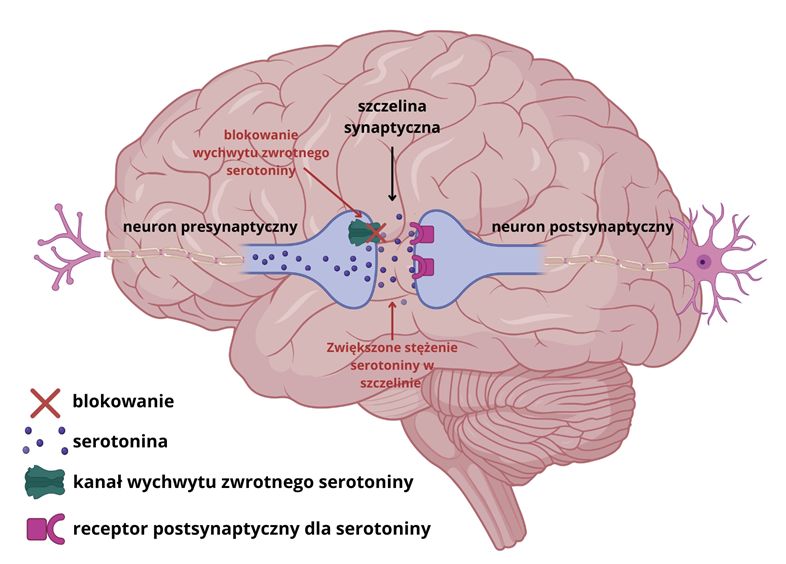
Hipoteza zakłada, że przyczyną depresji jest niedobór neuroprzekaźników w mózgu, a mechanizm działania leków polega na podnoszeniu poziomu serotoniny (5-HT) i noradrenaliny (NA) w szczelinie synaptycznej, poprzez hamowanie ich wychwytu zwrotnego lub metabolizmu. Badania wspierają tę teorię, wskazując, że depresja może być związana z niedoborem tzw. monoamin, czyli substancji chemicznych w mózgu, takich jak serotonina, noradrenalina i dopamina. U osób z depresją zaobserwowano między innymi:
● Nawrót choroby, gdy organizm nie produkuje wystarczającej ilości ważnych substancji, np. tryptofanu, który jest potrzebny do wytwarzania serotoniny, dlatego bardzo często w depresji zaleca się suplementację tryptofanu.
● Częstsze mutacje w genie odpowiedzialnym za produkcję enzymu (TPH-2), który pomaga wytwarzać serotoninę w mózgu.
● Zwiększoną aktywność enzymu MAO-A, który rozkłada neuroprzekaźniki (serotonina, noradrenalina), co może prowadzić do ich niedoboru.
● Nieprawidłową pracę receptorów serotoninowych (5-HT1A i 5-HT1B), które odpowiadają za prawidłowe przekazywanie sygnałów między komórkami nerwowymi, a w efekcie za nasze samopoczucie.
● Niski poziom białka p11, które wpływa na działanie serotoniny.
● Nieprawidłową reakcję białek G, które pomagają komórkom nerwowym odbierać sygnały chemiczne.
● Zmniejszone ilości substancji takich jak cAMP i inozytol, które biorą udział w przekazywaniu sygnałów w mózgu.
● Obniżony poziom białka CREB, które jest związane z regulacją nastroju – stwierdzono to w badaniach mózgów osób, które popełniły samobójstwo.
Wszystkie te zmiany sugerują, że depresja może być związana z zaburzeniami w produkcji i działaniu substancji chemicznych odpowiedzialnych za prawidłowe funkcjonowanie mózgu.
Kontrargumentami dla tej hipotezy są:
● Leki przeciwdepresyjne (LPD) muszą być stosowane przez dłuższy czas, aby przyniosły efekt terapeutyczny, co sugeruje, że samo podniesienie poziomu neuroprzekaźników nie wystarcza do szybkiego wyleczenia depresji.
● Obniżenie poziomu serotoniny (5-HT) lub noradrenaliny (NA) u zdrowych osób nie powoduje depresji, co podważa teorię, że ich niedobór jest bezpośrednią przyczyną choroby.
● Substancje zwiększające poziom monoamin, takie jak amfetamina czy kokaina, nie mają działania przeciwdepresyjnego, co sugeruje, że sama obecność tych neuroprzekaźników nie wystarcza do poprawy nastroju w depresji.
● Istnieją skuteczne leki przeciwdepresyjne, które nie wpływają na poziom monoamin, lecz działają poprzez blokowanie określonych receptorów w mózgu, co wskazuje na bardziej złożony mechanizm depresji.
2. HIPOTEZA ZMIAN ADAPTACYJNYCH
● Leki przeciwdepresyjne zaczynają działać dopiero po 2-3 tygodniach, mimo że poziom neuroprzekaźników wzrasta wcześniej. To sugeruje, że sama ich ilość nie wystarcza do leczenia depresji.
● Długotrwałe stosowanie leków powoduje zmiany adaptacyjne w mózgu, których nie obserwuje się po jednorazowym podaniu. Dochodzi do adaptacji receptorów nerwowych do nowych warunków.
● Mózg reaguje na leczenie, zmieniając wrażliwość receptorów, co może być kluczowe dla skuteczności terapii – nie sama ilość neuroprzekaźników, ale to, jak organizm je wykorzystuje, ma znaczenie.
Zmiany adaptacyjne w układzie noradrenergicznym (NA)
● Leki przeciwdepresyjne powodują zmiany w układzie nerwowym, dostosowując jego reakcje na bodźce. Jednym z pierwszych odkrytych efektów była tzw. "down"-regulacja receptorów β-adrenergicznych, czyli zmniejszenie ich liczby i wrażliwości, co jest związane z działaniem większości leków przeciwdepresyjnych oraz elektrowstrząsów.
● Niektóre leki nasilają działanie receptorów α1-adrenergicznych - "up"-regulacja, co wpływa na pobudzenie układu nerwowego i może być jednym z mechanizmów ich skuteczności.
● Zmniejsza się też wrażliwość receptorów α2-adrenergicznych, które normalnie ograniczają uwalnianie serotoniny i noradrenaliny – ich osłabienie prowadzi do zwiększenia ilości tych substancji w mózgu, co może poprawiać nastrój.
● Interakcje między receptorami α i β mogą dodatkowo wzmacniać działanie leków przeciwdepresyjnych, mimo że na poziomie pojedynczych receptorów zachodzą pozornie sprzeczne zmiany.
Zmiany adaptacyjne w układzie serotoninergicznym (5HT)
● Wpływ leków przeciwdepresyjnych na receptory serotoninowe (5-HT1 i 5-HT2) nie jest jednoznaczny – różne badania pokazują odmienne wyniki.
● Większość leków przeciwdepresyjnych osłabia działanie receptorów 5-HT2A, natomiast elektrowstrząsy mają odwrotny efekt.
● W przypadku receptorów 5-HT1A wyniki są sprzeczne – niektóre badania pokazują ich wzrost, inne spadek lub brak zmian. Może to wynikać z ich różnych funkcji:
- Presynaptyczne receptory 5-HT1A hamują aktywność neuronów serotoninowych, zatem ich osłabienie przez SSRI i inhibitory MAO może zwiększać ilość serotoniny w mózgu.
- Postsynaptyczne receptory 5-HT1A wzmacniają działanie serotoniny, a elektrowstrząsy dodatkowo je aktywują.
● Poziom receptorów 5-HT2A jest wyższy w mózgach osób, które popełniły samobójstwo, co wskazuje na związek zaburzeń funkcjonowania układu serotoninowego z depresją i samobójstwami. Potwierdza to raz jeszcze założenie, że depresja jest chorobą fizjologiczną, która nieleczona prowadzi do tragicznych skutków.
Zmiany adaptacyjne w układzie dopaminergicznym (DA)
● Leki przeciwdepresyjne nie wpływają na układ dopaminowy po jednorazowym podaniu, ale ich długotrwałe stosowanie prowadzi do zmian adaptacyjnych.
● Dochodzi do osłabienia receptorów D1 - "down"-regulacja, co oznacza zmniejszenie ich liczby.
● Jednocześnie wzmacniane są receptory D2 "up"-regulacja, co zwiększa ich wrażliwość na dopaminę.
● Podobne zmiany zachodzą także w przypadku receptorów D3 – mogą one odgrywać rolę w działaniu leków przeciwdepresyjnych.
3. HIPOTEZA PRZEWLEKŁEGO STRESU
Stres jest jedną z głównych przyczyn depresji – prowadzi do zmian w mózgu, zwłaszcza w hipokampie i korze czołowej.
Mechanizm stresu:
● Stres aktywuje tzw. oś HPA (podwzgórze–przysadka–nadnercza).
● Podwzgórze wydziela kortykoliberynę (CRH), która pobudza przysadkę do produkcji adrenokortykotropiny (ACTH).
● ACTH stymuluje nadnercza do wydzielania kortyzolu – głównego hormonu stresu.
● W normalnych warunkach kortyzol działa na zasadzie sprzężenia zwrotnego, hamując nadmierną aktywność osi HPA.
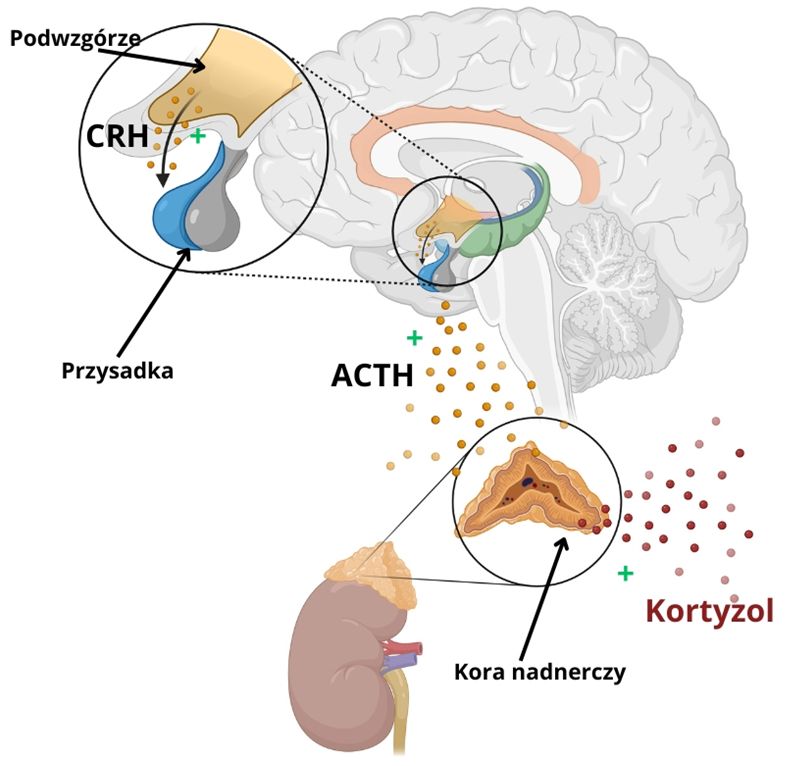
W depresji ten mechanizm jest zaburzony:
● Nadmiar kortyzolu prowadzi do uszkodzenia neuronów w hipokampie i korze czołowej, co wpływa na emocje i nasila depresję.
● Zmniejsza się objętość hipokampa i liczba połączeń nerwowych, co potwierdzają badania mózgów osób z depresją.
● Wysoki poziom kortyzolu może również hamować neurogenezę (powstawanie nowych neuronów).
U pacjentów z depresją często obserwuje się:
● Podwyższony poziom kortyzolu we krwi.
● Nadmiar CRF w płynie mózgowo-rdzeniowym.
● Nieprawidłową reakcję organizmu na stres.
● Długotrwały stres w dzieciństwie może trwale uszkodzić oś HPA, zwiększając ryzyko depresji w dorosłym życiu.
4. HIPOTEZA TROFICZNA
Neurotroficzna hipoteza depresji zakłada, że niski poziom neurotrofin, takich jak BDNF (brain-derived neurotrophic factor), przyczynia się do atrofii hipokampa i rozwoju depresji.
Rola BDNF:
● Jest kluczowy dla przeżycia neuronów, wzrostu aksonów i plastyczności synaptycznej.
● Jego poziom spada pod wpływem stresu i kortyzolu.
● U pacjentów z depresją, zwłaszcza po samobójstwie, obserwuje się obniżony poziom BDNF w hipokampie.
● Leki przeciwdepresyjne (LPD) podnoszą poziom BDNF, co może przyczyniać się do odbudowy uszkodzonych neuronów i zmniejszenia objawów depresji.
Mechanizm działania LPD:
● Antydepresyjne działanie BDNF pośredniczy poprzez czynnik transkrypcyjny CREB, który aktywuje jego ekspresję.
● LPD zwiększają poziom CREB w hipokampie i korze mózgowej.
● Czas potrzebny na wzrost poziomu BDNF może tłumaczyć opóźnioną skuteczność leków przeciwdepresyjnych.
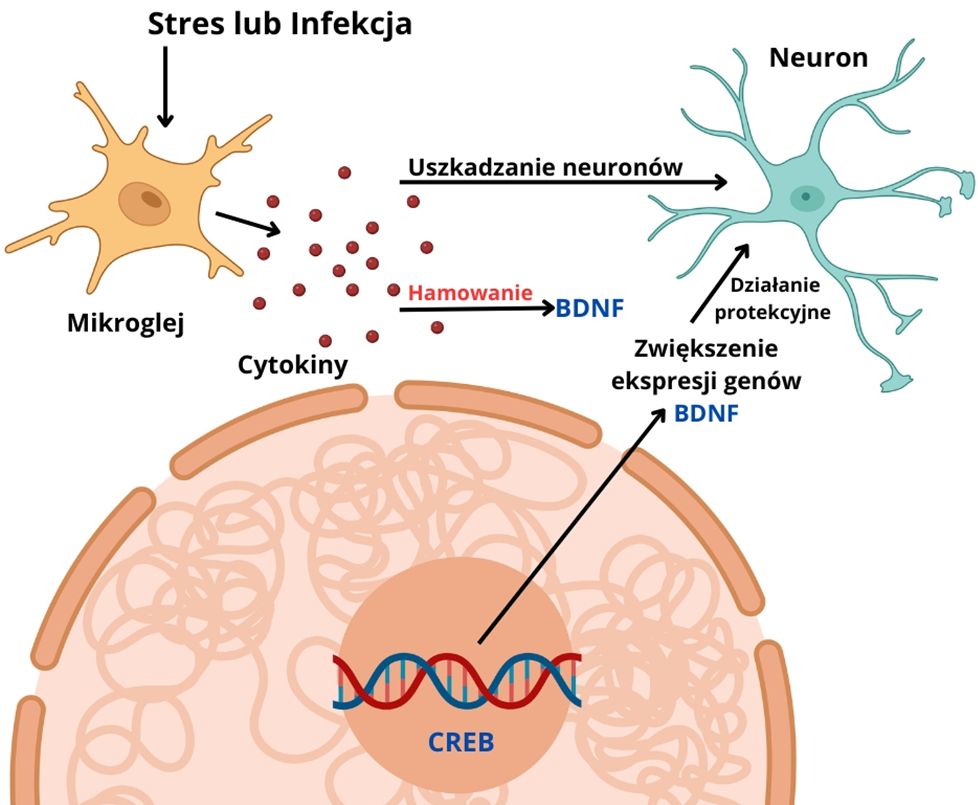
Ćwiczenia fizyczne i dieta mogą działać podobnie do LPD, zwiększając poziom BDNF i wspierając neurogenezę.
Depresja i choroby sercowo-naczyniowe mogą mieć wspólne mechanizmy, związane z zaburzoną funkcją śródbłonka i niedoborem BDNF.
Choć neurotroficzna hipoteza nie doprowadziła do powstania nowych leków, znacząco poszerzyła wiedzę o mechanizmach depresji i roli neurogenezy w jej leczeniu.
5. HIPOTEZA ZABURZEŃ NEUROPLASTYCZNOŚCI ORAZ HIPOTEZA SIECI NEURONALNEJ
Depresja a zmiany neuroplastyczne:
● Zaburzenia neuroplastyczności w obszarach kontrolujących nastrój prowadzą do objawów depresyjnych.
● Badania na zwierzętach i analizy postmortem wykazały:
- Osłabienie neurogenezy w hipokampie u dorosłych.
- Zmniejszenie gęstości komórek glejowych, utratę aksonów i oznaki martwicy neuronów w korze przedczołowej.
- Demielinizację oraz wzmożone przekaźnictwo synaptyczne.
● Zmiany morfologiczne w mózgach osób z depresją:
- Zmniejszenie objętości istoty szarej w korze przedczołowej (przyśrodkowej i oczodołowej).
- Atrofia hipokampa i brzusznej części prążkowia.
- Poszerzenie komory trzeciej.
- Zwiększony przepływ krwi w jądrze migdałowatym, korelujący z nasileniem objawów depresji, co wpływa na bardziej emocjonalne zachowania.
● Neuroplastyczność a działanie leków przeciwdepresyjnych (LPD):
- LPD działają nie tylko na poziomie synaps, ale również aktywują procesy adaptacyjne:
- Aktywacja kaskady cAMP.
- Wzrost czynników neurotroficznych (np. BDNF).
- Nasilenie neurogenezy i synaptogenezy.
● Odbudowa sieci neuronalnych przywraca prawidłowe funkcjonowanie obwodów mózgowych związanych z nastrojem.

U osoby chorej obserwujemy ubytek istoty szarej kory mózgowej, co prowadzi do osłabienia kontroli nad ruchem, pamięcią, emocjami oraz innymi kluczowymi funkcjami niezbędnymi w codziennym życiu. Może to skutkować trudnościami w podejmowaniu decyzji, regulacji nastroju oraz przetwarzaniu informacji, co dodatkowo pogłębia objawy choroby. Grafika po lewej stronie przedstawia nadaktywne ciało migdałowate, które odgrywa kluczową rolę w regulacji emocji, reakcji na stres oraz funkcjonowaniu układu odpornościowego. Jego nadmierna aktywność może prowadzić do zwiększonej podatności na lęk, nadmiernej reakcji na bodźce stresowe oraz trudności w kontrolowaniu emocji, co jest charakterystyczne dla depresji i innych zaburzeń nastroju.
Teoria sieci neuronalnych:
● Zaburzenia nastroju wynikają z dysfunkcji krytycznych sieci limbiczno-wzgórzowo-korowych i limbiczno-prążkowiowo-gałkowo-wzgórzowych.
● Czynniki neurotroficzne nie kontrolują nastroju bezpośrednio, lecz umożliwiają plastyczność neuronalną, niezbędną do odbudowy tych sieci.
● LPD stymulują lokalną aktywność neuronalną, co sprzyja neurogenezie, synaptogenezie i dojrzewaniu neuronów.
Opóźniony efekt działania LPD:
● Ich skuteczność wiąże się z procesami neuroplastycznymi, które wymagają czasu na odbudowę sieci neuronalnych.
● Elektrowstrząsy i LPD pobudzają neurogenezę w hipokampie, co potwierdzają badania kliniczne.
Podsumowując, hipoteza neuroplastyczności tłumaczy depresję jako skutek zaburzeń adaptacyjnych w mózgu, a skuteczność leczenia jako odbudowę funkcjonalnych połączeń neuronalnych.
ROLA UKŁADU GLUTAMINERGICZNEGO W PATOMECHANIZMIE DEPRESJI
Receptory NMDA odgrywają kluczową rolę w przekaźnictwie sygnałów w mózgu i wpływają na różne procesy, takie jak uczenie się, pamięć oraz adaptacja, ale są też związane z poważnymi chorobami, w tym depresją. Oto najważniejsze punkty dotyczące receptorów NMDA i ich wpływu na leczenie depresji:
1. Rola układu glutaminianergicznego:
● W mózgu istnieją dwa główne systemy:
- pobudzający (glutaminianergiczny) i
- hamujący (GABA-ergiczny), które pomagają utrzymać równowagę.
Glutaminian (Glu) jest głównym przekaźnikiem sygnałów w układzie glutaminianergiczny, a receptory NMDA są kluczowe dla jego działania.
2. Budowa i funkcja receptorów NMDA:
Receptory NMDA są zbudowane z różnych podjednostek i mają kanały, które przepuszczają jony wapnia (Ca²⁺), sód (Na⁺) i potas (K⁺), co wpływa na aktywność neuronów. Te receptory są obecne w ważnych częściach mózgu, takich jak hipokamp i kora mózgowa.
3. Modulacja receptora NMDA:
Aktywacja receptora NMDA wymaga obecności dwóch agonistów: glutaminianu i glicyny. Dopiero po ich związaniu receptor pozwala na przepływ jonów. Kanał NMDA jest blokowany przez magnez, który zostaje usunięty dopiero po depolaryzacji błony komórkowej.
4. Związek z plastycznością mózgu:
Receptory NMDA są niezbędne dla plastyczności synaptycznej, co oznacza, że pomagają w procesach zapamiętywania i uczenia się. Dysfunkcja tych receptorów może przyczyniać się do zaburzeń, w tym depresji.
5. Depresja i receptory NMDA:
Zarówno zmniejszona aktywność receptorów NMDA jak i ich przewlekłe pobudzenie może prowadzić do depresji. Stymulowanie tych receptorów może poprawiać nastrój, dlatego leki, które modulują te receptory (np. ketamina), okazały się skuteczne w leczeniu depresji, zwłaszcza w przypadkach opornych na tradycyjne terapie.
6. Ketamina i inne leki:
Ketamina, działająca jako antagonista receptora NMDA, ma szybki efekt przeciwdepresyjny, co sugeruje, że blokada tych receptorów może przynieść korzyści w leczeniu depresji. Jednak jej długoterminowe efekty i bezpieczeństwo wymagają dalszych badań.
Podsumowując, receptory NMDA odgrywają istotną rolę w regulacji funkcji mózgu, a ich modulacja stanowi nowoczesne podejście w leczeniu depresji, zwłaszcza w przypadkach, gdzie inne terapie zawodzą.
● Rola glutaminianu: U osób z depresją obserwuje się podwyższony poziom glutaminianu (głównego neuroprzekaźnika pobudzającego) w surowicy krwi, płytkach krwi i płynie mózgowo-rdzeniowym.
● Receptor NMDA: Nadmierna aktywacja tego receptora może prowadzić do uszkodzeń neuronów, zaburzeń pamięci i zwiększonej podatności na stres.
● Zmiany w mózgu: U osób z depresją i ofiar samobójstw wykazano zaburzenia w funkcjonowaniu receptora NMDA, szczególnie w hipokampie i korze czołowej.
● Nadmierna aktywność receptora NMDA jest związana z negatywnymi skutkami stresu, jak:
- Zanik neuronów.
- Osłabienie neurogenezy (tworzenia nowych komórek nerwowych).
- Zjawisko kindling (rozniecanie) – zwiększona wrażliwość układu nerwowego na bodźce.
- Zjawisko ekscytotoksyczności - śmierć komórek nerwowych przez glutaminian
● Blokowanie receptora NMDA może odwrócić te procesy i wykazuje działanie przeciwdepresyjne.
● Ketamina:
- Działa szybko – poprawia nastrój nawet w ciągu kilku godzin.
- Szczególnie skuteczna w depresji opornej na leczenie.
- Efekt utrzymuje się do dwóch tygodni po jednorazowym podaniu.
- Działa poprzez przesunięcie aktywności z receptorów NMDA na AMPA, co stymuluje neuroplastyczność (zdolność mózgu do regeneracji).
● Inne leki (np. memantyna, amantadyna):
- Mają łagodniejszy wpływ na receptor NMDA i mniej skutków ubocznych.
- Trwają badania nad selektywnymi antagonistami, które mogą być bezpieczniejsze i skuteczniejsze.
● Skutki uboczne, takie jak:
- Objawy psychotyczne (np. halucynacje).
- Zaburzenia pamięci.
- Ryzyko uzależnienia.
- Potencjalna neurotoksyczność.
● Dlatego badania koncentrują się na lekach o bardziej precyzyjnym działaniu.
● Selektywne blokowanie podjednostek NR2B:
- Mniej skutków ubocznych.
- Obiecujące wyniki w leczeniu depresji lekoopornej (np. CP-101,606).
● Blokada syntezy tlenku azotu (NO):
- Hamowanie NO zmniejsza toksyczny wpływ aktywacji NMDA.
- Związki takie jak błękit metylenowy wykazują działanie przeciwdepresyjne.
● Aktywacja receptorów AMPA (AMPAkiny):
- Stymulują produkcję BDNF (białka wspierającego wzrost neuronów).
- Współdziałają z antagonistami NMDA w łagodzeniu objawów depresji.
● Długotrwałe stosowanie leków przeciwdepresyjnych (LPD) prowadzi do:
- Osłabienia funkcji receptorów NMDA.
- Wzrostu aktywności receptorów AMPA.
- Zmian w proporcjach podjednostek receptorów.
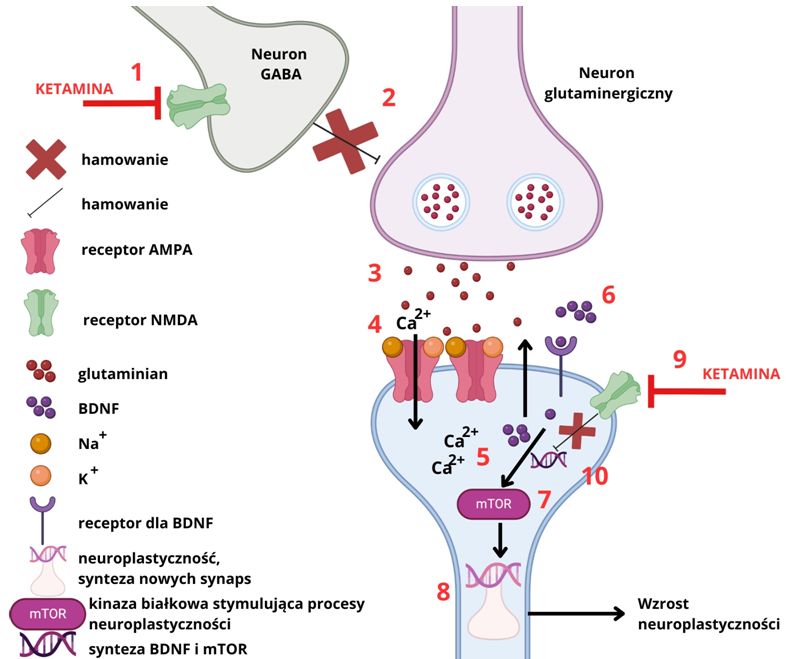
1. Ketamina blokuje receptor NMDA na neuronie GABAergicznym, który jest neuronem hamującym.
2. Dochodzi do inhibicji wpływu hamującego neuronu GABA na neuron glutaminergiczny.
3. W efekcie odhamowania neuronu glutaminergicznego do szczeliny synaptycznej uwalniany jest glutaminian,który kolejno stymuluje receptory AMPA.
4. Do wnętrza komórki przewodzone są jony Ca2+.
5. Wzrost stężenia jonów wapniowych w komórce powoduje uwolnienie czynnika BDNF do szczeliny synaptycznej.
6. BDNF wiąże się ze swoim receptorem powierzchniowym i go aktywuje co prowadzi do kolejnej kaskady reakcji wewnątrz neuronu.
7. Aktywowana jest kinaza białkowa mTOR, która kolejno pobudza procesy syntezy białek.
8. Te nowo syntetyzowane białka są następnie wprowadzane do gęstości postsynaptycznej, co prowadzi do dalszego wzrostu transmisji synaptycznej pośredniczonej przez AMPAR i gęstości kolców dendrytycznych, powodując w ten sposób masywną synaptogenezę.
9. Ketamina hamuje wpływ hamujący receptorów NMDA na syntezę czynnika BDNF oraz kinazy mTOR, przez co zapobiega śmierci neuronu i umożliwia procesy neuroplastyczności.
● Czym są receptory metabotropowe?
Receptory metabotropowe (mGluR) to białka związane z neuroprzekaźnikiem glutaminianem, które regulują przekazywanie sygnałów w mózgu i wpływają na metabolizm komórkowy.
● Jak działają?
- Współpracują z białkami G, modulując poziom różnych przekaźników w neuronach.
- Regulują aktywność mózgu poprzez pobudzenie lub hamowanie komórek nerwowych.
● Podział receptorów mGluR
Receptory te dzielą się na trzy grupy, różniące się lokalizacją i funkcją:
1. Grupa I (mGluR1, mGluR5) – pobudzają aktywność neuronów, wpływają na regulację nastroju i funkcje poznawcze.
2. Grupa II (mGluR2, mGluR3) – hamują nadmierne pobudzenie, mogą regulować uwalnianie neuroprzekaźników takich jak GABA.
3. Grupa III (mGluR4, mGluR6, mGluR7, mGluR8) – wpływają na dostępność glutaminianu oraz innych neuroprzekaźników, co może mieć znaczenie w kontroli emocji i zachowania.
● Dlaczego są ważne?
- Uczestniczą w procesach związanych z pamięcią, emocjami i plastycznością mózgu.
- Ich nieprawidłowe funkcjonowanie może mieć wpływ na depresję, lęk i inne zaburzenia neurologiczne.
- Stanowią potencjalny cel dla nowych terapii neurologicznych i psychiatrycznych.
● Nadmierna aktywacja receptorów NMDA w mózgu jest związana z depresją.
● Antagoniści NMDA (zwłaszcza ketamina) szybko łagodzą objawy depresji, ale mogą powodować skutki uboczne.
● Nowe terapie skupiają się na bardziej precyzyjnym blokowaniu receptorów NMDA lub modulacji receptorów AMPA i metabotropowych, co może zapewnić skuteczność przy mniejszym ryzyku działań niepożądanych.
Podsumowanie problemu depresji
Podsumowując, depresja to bardzo złożona choroba, której przyczyny mogą być różnorodne i obejmować szereg mechanizmów biologicznych, psychicznych oraz środowiskowych. Każdy przypadek depresji jest inny, a jej leczenie wymaga indywidualnego podejścia, które łączy psychoterapię z odpowiednio dobranym leczeniem farmakologicznym. To schorzenie, z którego nie można wyjść w krótkim czasie, ponieważ nie zależy wyłącznie od naszej woli, lecz również od funkcjonowania naszego mózgu i jego reakcji na stres, zmiany hormonalne czy inne czynniki.
Warto zadbać o siebie i swoje zdrowie psychiczne, szukając pomocy, gdy jej potrzebujemy. Nie bójmy się sięgać po leki, pójść do psychiatry czy psychoterapeuty – są oni po to, by pomóc nam lepiej radzić sobie z trudnościami, zrozumieć siebie i odzyskać równowagę. Współczesna medycyna oferuje skuteczne narzędzia, które mogą poprawić jakość życia i pomóc wrócić do pełnej sprawności psychicznej.
Nie pozwólmy, by krzywdzące stereotypy i stygmatyzacja blokowały nas w poszukiwaniach pomocy. Depresja to problem, który dotyka ogromną część społeczeństwa i nie ma związku z wiekiem, statusem społecznym czy siłą charakteru. Każdy z nas może znaleźć się w trudnej sytuacji życiowej, której skutkiem będą zmiany w organizmie, emocjach i postrzeganiu rzeczywistości. Dlatego nie oceniajmy, nie wytykajmy palcem, a zamiast tego bądźmy wsparciem – zarówno dla siebie, jak i dla innych.
Dbajmy o zdrowie psychiczne, bo to równie ważne, jak troska o zdrowie fizyczne. Warto pamiętać, że z pomocą, wsparciem i odpowiednim leczeniem depresja jest chorobą, z której można wyjść i odzyskać kontrolę nad własnym życiem.
Autor artykułu : Magister Farmacji Klaudia Kowalczyk
Bibliografia
1. Światowa Organizacja Zdrowia (WHO). (2017). Depresja: porozmawiajmy o niej. https://www.who.int/news-room/fact-sheets/detail/depression
2. Narodowy Fundusz Zdrowia (NFZ). (2021). Raport o stanie zdrowia dzieci i młodzieży w Polsce. https://www.nfz.gov.pl/aktualnosci/aktualnosci-centrali/raport-o-stanie-zdrowia-dzieci-i-mlodziezy-w-polsce,7410.html
3. Niewidoczna Pomoc. (2023). Depresja u dzieci i młodzieży: statystyki, rodzaje, przyczyny i objawy. https://niewidacpomnie.org/2023/02/20/depresja-dzieci-i-mlodziezy-statystyka-rodzaje-przyczyny-i-objawy-depresji-u-dzieci/
4. Schildkraut, J. J. (1965). The catecholamine hypothesis of affective disorders: a review of supporting evidence. American Journal of Psychiatry, 122(5), 509–522. https://doi.org/10.1176/ajp.122.5.509
5. Coppen, A. (1967). The biochemistry of affective disorders. British Medical Bulletin, 23(3), 309–318. https://doi.org/10.1093/oxfordjournals.bmb.a070358
6. Nowakowska, I., et al. (2005). The role of serotonin in the pathophysiology of depression. Polish Journal of Pharmacology, 57(3), 227–234. https://pubmed.ncbi.nlm.nih.gov/15863556/
7. Pilc, A., et al. (2000). The role of serotonin in the pathophysiology of depression. Polish Journal of Pharmacology, 52(5), 413–420. https://pubmed.ncbi.nlm.nih.gov/11197356/
8. Belmaker, R. H., et al. (2008). Monoamine oxidase inhibitors in the treatment of depression. Journal of Clinical Psychiatry, 69(3), 341–348. https://doi.org/10.4088/JCP.v69n0303
9. Elhwuegi, A. S. (2004). Central monoamines and their role in major depression. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 28(3), 435–451. https://doi.org/10.1016/j.pnpbp.2003.10.016
10. Ruhé, H. G., et al. (2007). The serotonin transporter gene and antidepressant efficacy: a systematic review and meta-analysis. European Neuropsychopharmacology, 17(2), 96–105. https://doi.org/10.1016/j.euroneuro.2006.07.004
11. Zhang, X., et al. (2005). Association between the TPH2 gene and major depressive disorder in a Chinese population. Psychiatric Genetics, 15(5), 311–314. https://doi.org/10.1097/01.ypg.0000189070.35847.3b
12. Meyer, J. H., et al. (2006). Elevated monoamine oxidase a levels in the brain: an explanation for the monoamine imbalance of major depression. Archives of General Psychiatry, 63(11), 1209–1216. https://doi.org/10.1001/archpsyc.63.11.1209
13. Pitchot, W., et al. (2005). Serotonergic dysfunction in depression: evidence from studies of platelet 5-HT2A receptor binding. Journal of Affective Disorders, 85(3), 271–277. https://doi.org/10.1016/j.jad.2004.12.010
14. Svenningsson, P., et al. (2006). Alterations in 5-HT1A receptor function and expression in depression: studies in human and non-human primates. European Journal of Pharmacology, 535(1–3), 72–77. https://doi.org/10.1016/j.ejphar.2005.12.029
15. Chang, L. H., et al. (2002). G protein-coupled receptor kinase 2 (GRK2) in the brain: a potential target for antidepressant therapy. Journal of Neuroscience Research, 68(5), 601–607. https://doi.org/10.1002/jnr.10247
16. Avissar, S., et al. (1997). A review of the mechanisms of action of antidepressant drugs. Journal of Clinical Psychiatry, 58(7), 17-25.
17. Valdizán, E. M., et al. (2003). Altered signal transduction pathways in the brain in depression. European Neuropsychopharmacology, 13(1), 1-6.
18. Blendy, J. A. (2006). The CREB family of transcription factors in addiction and depression. The European Journal of Pharmacology, 582(1), 28–34.
19. Kessler, R. C., et al. (2003). Depression and anxiety disorders in the World Health Organization World Mental Health Surveys. World Psychiatry, 2(3), 121-128.
20. Beck, A. T., & Alford, B. A. (2009). Depression: Causes and treatment. University of Pennsylvania Press.
21. American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders: DSM-5 (5th ed.). American Psychiatric Publishing.
22. Nolen-Hoeksema, S. (2014). Emotion regulation and psychopathology: The role of cognitive emotion regulation strategies. Annual Review of Clinical Psychology, 10, 17-42.
23. Tiller, J. W. (2013). Depression and anxiety. Medical Journal of Australia, 199(6), 28-32.
24. Möller, H. J., et al. (2008). The neurobiology of major depression. Current Opinion in Psychiatry, 21(1), 34-42.
25. Boulton, M., et al. (2005). Cognitive theories of depression: A review of the evidence for the cognitive model of depression. Clinical Psychology Review, 25(7), 964-981.
26. Fava, G. A., et al. (2015). Psychotherapeutic approaches to depression and anxiety. International Review of Psychiatry, 27(3), 221-231.
27. Kendler, K. S., et al. (2002). Genetic and environmental influences on the risk of major depression. American Journal of Psychiatry, 159(10), 1706-1712.
28. Borsini, F., & Meli, A. (1999). Antidepressant-like activity in animal models: A review of methods and mechanisms. Neuroscience and Biobehavioral Reviews, 23(2), 265-280.
29. Harmer, C. J., et al. (2009). The effects of acute antidepressant administration on emotion processing: Implications for mechanisms of therapeutic change. European Neuropsychopharmacology, 19(7), 441-452.
30. </
